This resource is a video abstract of a research paper created by …
This resource is a video abstract of a research paper created by Research Square on behalf of its authors. It provides a synopsis that's easy to understand, and can be used to introduce the topics it covers to students, researchers, and the general public. The video's transcript is also provided in full, with a portion provided below for preview:
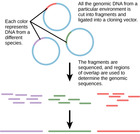
"“All happy families look alike; each unhappy family is unhappy in its own way” – Leo Tolstoy, Anna Karenina. According to Tolstoy, a healthy home is the result of many factors falling into harmonious order, whereas disharmony is what happens when even one of these factors is out of place. A new study confirms the same principle holds true for the communities of microbes that determine oral health. Researchers mapped microbial DNA from healthy individuals and individuals with one of three forms of gum infection: chronic periodontitis, localized aggressive periodontitis, or generalized aggressive periodontitis. While it’s known that all three forms of periodontitis are microbially derived, the microbial makeup that gives rise to each remains unclear. High-throughput whole genome sequencing revealed that, like Tolstoy's unhappy homes, no two individuals with disease were alike..."
The rest of the transcript, along with a link to the research itself, is available on the resource itself.
This resource is a video abstract of a research paper created by …
This resource is a video abstract of a research paper created by Research Square on behalf of its authors. It provides a synopsis that's easy to understand, and can be used to introduce the topics it covers to students, researchers, and the general public. The video's transcript is also provided in full, with a portion provided below for preview:
"Metagenomic analysis frequently plays an important role in development pipelines for human fecal microbiome-related products, but validation and standardization of the methods used to extract DNA and assemble sequence libraries for these studies is currently lacking. To close this gap, researchers recently characterized existing protocols for accuracy and precision. First, they tested the quantification accuracy by using a defined mock community of bacteria. Then, the protocols that performed as expected were evaluated for both within- and inter-laboratory precision metrics. The protocols were also tested against the MOSAIC Standards Challenge samples. Lastly, they defined performance metrics for the recommended protocols to provide best-practice guidance. The uptake of the recommendations generated here should improve reproducibility in human metagenomic research and therefore facilitate development and commercialization of human microbiome-related products..."
The rest of the transcript, along with a link to the research itself, is available on the resource itself.
Biology is designed for multi-semester biology courses for science majors. It is …
Biology is designed for multi-semester biology courses for science majors. It is grounded on an evolutionary basis and includes exciting features that highlight careers in the biological sciences and everyday applications of the concepts at hand. To meet the needs of today’s instructors and students, some content has been strategically condensed while maintaining the overall scope and coverage of traditional texts for this course. Instructors can customize the book, adapting it to the approach that works best in their classroom. Biology also includes an innovative art program that incorporates critical thinking and clicker questions to help students understand—and apply—key concepts.
By the end of this section, you will be able to:Describe gel …
By the end of this section, you will be able to:Describe gel electrophoresisExplain molecular and reproductive cloningDescribe uses of biotechnology in medicine and agriculture
Biology is designed for multi-semester biology courses for science majors. It is …
Biology is designed for multi-semester biology courses for science majors. It is grounded on an evolutionary basis and includes exciting features that highlight careers in the biological sciences and everyday applications of the concepts at hand. To meet the needs of today’s instructors and students, some content has been strategically condensed while maintaining the overall scope and coverage of traditional texts for this course. Instructors can customize the book, adapting it to the approach that works best in their classroom. Biology also includes an innovative art program that incorporates critical thinking and clicker questions to help students understand—and apply—key concepts.
This course teaches the design of contemporary information systems for biological and …
This course teaches the design of contemporary information systems for biological and medical data. Examples are chosen from biology and medicine to illustrate complete life cycle information systems, beginning with data acquisition, following to data storage and finally to retrieval and analysis. Design of appropriate databases, client-server strategies, data interchange protocols, and computational modeling architectures. Students are expected to have some familiarity with scientific application software and a basic understanding of at least one contemporary programming language (e.g. C, C++, Java, Lisp, Perl, Python). A major term project is required of all students. This subject is open to motivated seniors having a strong interest in biomedical engineering and information system design with the ability to carry out a significant independent project. This course was offered as part of the Singapore-MIT Alliance (SMA) program as course number SMA 5304.
This resource is a video abstract of a research paper created by …
This resource is a video abstract of a research paper created by Research Square on behalf of its authors. It provides a synopsis that's easy to understand, and can be used to introduce the topics it covers to students, researchers, and the general public. The video's transcript is also provided in full, with a portion provided below for preview:
"The development of long-read sequencing has allowed for the generation of more complete and contiguous genomes in metagenomics studies. However, long-reads are more prone to sequencing errors than short-reads, and these errors can end up incorporated in the draft genomes. Combining short- and long-reads can overcome such errors, but is computationally taxing. To avoid this, researchers developed the ‘Hierarchical Clustering Based Hybrid Assembly (HCBHA) approach.’ This approach first groups the long- and short-reads into candidate bacterial haplotypes and then assembles each group separately, which reduces the computational demand . Researchers tested this framework on a microbiome from activated sludge, an important part of wastewater treatment. The highly complex microbiomes found in activated sludge remove pollutants from wastewater..."
The rest of the transcript, along with a link to the research itself, is available on the resource itself.
This resource is a video abstract of a research paper created by …
This resource is a video abstract of a research paper created by Research Square on behalf of its authors. It provides a synopsis that's easy to understand, and can be used to introduce the topics it covers to students, researchers, and the general public. The video's transcript is also provided in full, with a portion provided below for preview:
"High-quality reference genomes are needed to understand the physiology and function of uncultured microbes in complex ecosystems. Metagenomics has been an incredibly useful tool for studying microbial communities, but assigning sequence assemblies accurately to genomes is difficult in microbial species or strains that lack a reference genome. These 'consensus genomes' have lower resolution than those generated from cultured isolates. Combining single-cell genomics with metagenomics may allow us to overcome these methodological weaknesses. Thus, researchers recently developed a framework called SMAGLinker, which integrates single-cell genomes from microfluidic droplets and uses them as guides for metagenome assembly. Compared to metagenomics alone, SMAGLinker showed more precise contig binning and higher recovery rates of rRNA and plasmids in a mock microbial community. In human gut and skin microbiota samples, SMAGLinker returned more genomes than the conventional metagenomics frameworks..."
The rest of the transcript, along with a link to the research itself, is available on the resource itself.
This genomics education lesson plan was formulated and tested on some year …
This genomics education lesson plan was formulated and tested on some year 6 students with the help of their teacher Michelle Pardini at the Hong Kong ICS School. Using the example of the ongoing citizen science Bahinia Genome project from Hong Kong it hopes to serve as a model to inspire and inform other national genome projects, and aid the development of crucial genomic literacy and skills across the globe. Inspiring and training a new generation of scientists to use these tools to tackle the biggest threats to mankind: climate change, disease, and food security. It is released under a CC-BY SA 4.0 license, and utilised the following slide deck and final quiz. Promoting open science, all of the data and resources produced from the project is immediately put into the public domain. Please feel free to utilise, adapt and build upon any of these as you wish. The open licence makes these open education resources usable just with attribution and posting of modified resources under a similar manner. Contact BauhiniaGenome if you have any questions or feedback.Bauhinia Genome overviewFor a slidedeck for the lesson plan laid out here you can use the set in slideshare here.
This course covers the algorithmic and machine learning foundations of computational biology …
This course covers the algorithmic and machine learning foundations of computational biology combining theory with practice. We cover both foundational topics in computational biology, and current research frontiers. We study fundamental techniques, recent advances in the field, and work directly with current large-scale biological datasets.
With the growing availability and lowering costs of genotyping and personal genome …
With the growing availability and lowering costs of genotyping and personal genome sequencing, the focus has shifted from the ability to obtain the sequence to the ability to make sense of the resulting information. This course is aimed at exploring the computational challenges associated with interpreting how sequence differences between individuals lead to phenotypic differences in gene expression, disease predisposition, or response to treatment.
This course covers the analytical, graphical, and numerical methods supporting the analysis …
This course covers the analytical, graphical, and numerical methods supporting the analysis and design of integrated biological systems. Topics include modularity and abstraction in biological systems, mathematical encoding of detailed physical problems, numerical methods for solving the dynamics of continuous and discrete chemical systems, statistics and probability in dynamic systems, applied local and global optimization, simple feedback and control analysis, statistics and probability in pattern recognition. An official course Web site and Wiki is maintained on OpenWetWare: 20.181 Computation for Biological Engineers.
Data Carpentry's aim is to teach researchers basic concepts, skills, and tools …
Data Carpentry's aim is to teach researchers basic concepts, skills, and tools for working more effectively with data. The lessons below were designed for those interested in working with Genomics data in R.
Data Carpentry lesson to learn how to use command-line tools to perform …
Data Carpentry lesson to learn how to use command-line tools to perform quality control, align reads to a reference genome, and identify and visualize between-sample variation. A lot of genomics analysis is done using command-line tools for three reasons: 1) you will often be working with a large number of files, and working through the command-line rather than through a graphical user interface (GUI) allows you to automate repetitive tasks, 2) you will often need more compute power than is available on your personal computer, and connecting to and interacting with remote computers requires a command-line interface, and 3) you will often need to customize your analyses, and command-line tools often enable more customization than the corresponding GUI tools (if in fact a GUI tool even exists). In a previous lesson, you learned how to use the bash shell to interact with your computer through a command line interface. In this lesson, you will be applying this new knowledge to carry out a common genomics workflow - identifying variants among sequencing samples taken from multiple individuals within a population. We will be starting with a set of sequenced reads (.fastq files), performing some quality control steps, aligning those reads to a reference genome, and ending by identifying and visualizing variations among these samples. As you progress through this lesson, keep in mind that, even if you aren’t going to be doing this same workflow in your research, you will be learning some very important lessons about using command-line bioinformatic tools. What you learn here will enable you to use a variety of bioinformatic tools with confidence and greatly enhance your research efficiency and productivity.
No restrictions on your remixing, redistributing, or making derivative works. Give credit to the author, as required.
Your remixing, redistributing, or making derivatives works comes with some restrictions, including how it is shared.
Your redistributing comes with some restrictions. Do not remix or make derivative works.
Most restrictive license type. Prohibits most uses, sharing, and any changes.
Copyrighted materials, available under Fair Use and the TEACH Act for US-based educators, or other custom arrangements. Go to the resource provider to see their individual restrictions.